Theoretical and Computational Chemistry
Equipment
Aberdeen University operates its own in-house High Performance Computing cluster, “Maxwell” (named after James Clerk Maxwell , the eminent Scottish Theoretical Physicist who spent a number of years as a professor at the University's Marischal College). Maxwell is a Linux supercomputing cluster which provides: 1240 CPU cores and 12TB of RAM. The theoretical and computational chemistry group also makes use of national facilities like ARCHER-2 for larger scale calculations with time provided by the UK-AMOR High-End Computing (HEC) Consortium (of which Mark Law is a member).
Research
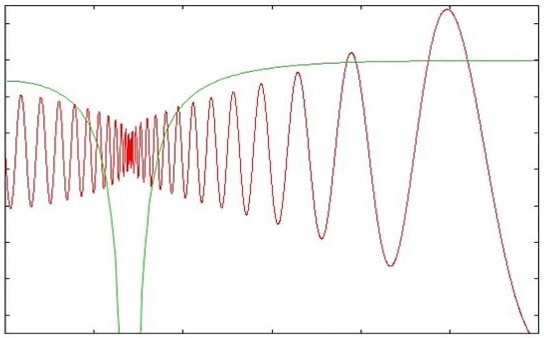
Isomerising molecules
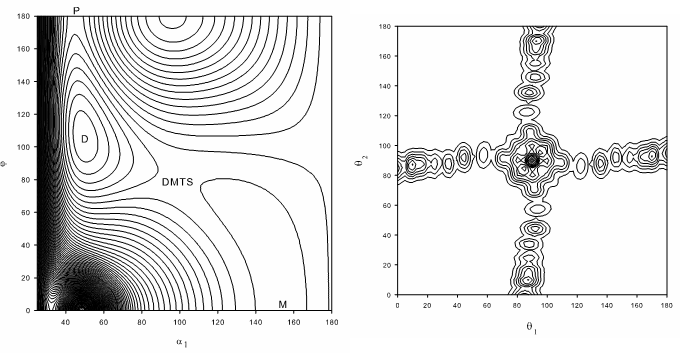
Interest in understanding wide-amplitude ('floppy') molecular motions has been stimulated by the drive to develop theories of intermolecular forces, isomerization and coherent control of chemical reactions. Methods for calculating the rotation-vibration energy levels and wavefunctions of floppy systems have advanced greatly but remain technically demanding and computationally expensive even for molecules and complexes as small as tetraatomics. We have studied the isovalent acetylene, silylidene and disilyne molecules which exhibit a surprising variety of isomeric structures (including some with bridging hydrogens). At sufficiently high (vibrational) energies, these molecules all undergo isomerisation (via 1,2 hydrogen shift reactions):
HCCH <-> H2CC
HCSiH <-> H2CSi <-> CSiH2
HSiSiH <-> H2SiSi <-> HSi(H)Si <-> Si(H)2Si
In each case we have calculated highly excited vibrational state energies and wavefunctions including for states delocalised over multiple isomeric forms.
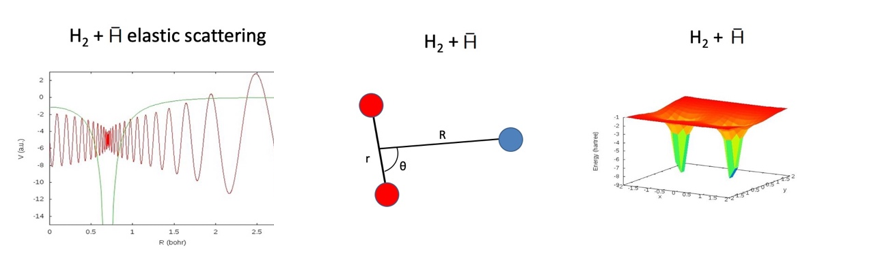
Antimatter interactions with normal matter
Antihydrogen atoms are now routinely created at CERN and can be stored for long enough to allow spectroscopy experiments to be carried out as very precise tests of their properties for comparison with ordinary matter. A major goal of these experimental efforts is to test fundamental symmetries (charge-parity-time, CPT) and potential physics beyond the Standard Model. This goal will require further significant developments in the experimental techniques over the next few years including in the accumulation of larger amounts of antihydrogen atoms and their storage over longer times (days or weeks). The latter is limited by the unavoidable presence of normal matter hydrogen molecules, H2, in the apparatus. Interactions of antihydrogen atoms with H2 lead to the destruction of the hard-won antimatter. Very little is known about these interactions but quantitative information will be crucial in guiding the experimental developments. Our theoretical research aims to overcome a major bottleneck in the understanding of matter-antimatter collisions by developing the capability to calculate the rates (or cross sections) for the various collision outcomes at the low energies relevant to the CERN experiments.
Activities
Funding
We have received funding from EPSRC, the Carnegie Trust, the Royal Society and the Nuffield Foundation.
Collaborations
We are always keen to engage with researchers within our fields of interest. Within the UK we have built strong connections with groups in St Andrews University, Durham University and University College London and further afield with Wrocław University of Science and Technology, Poland.
Interested in joining the group?
If you're interested in joining the group as a PhD student, please email Dr Mark Law, m.m.law@abdn.ac.uk
Selected Publications
- M. M. Law, J. T. Fraser-Smith and C. U. Perotto, "The potential energy surface of isomerising disilyne ", Physical Chemistry Chemical Physics 14, 6922 (2012).
- M. M. Law and C. U. Perotto, "The vibrational bound states of isomerising disilyne ", Journal of Chemical Physics, 139, 064308 (2013).
- B. P. Mant, M. M. Law and K. Strasburger, "Hydrogen molecule-antihydrogen atom potential energy surface and scattering calculations ", Journal of Physics B 52, 185201 (2019).