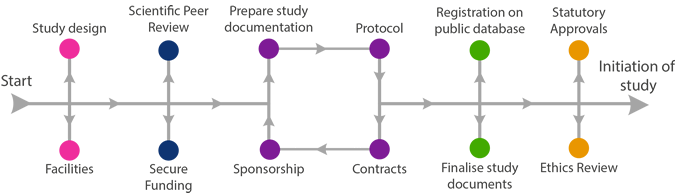
It is the single point of reference for the study for everyone involved, from the funders, who want to know about the project they are supporting, to the trial staff, patients and healthcare providers and finally to the journals when the paper is to be published.
In the case of drugs trials, the protocol becomes part of the EU Clinical Trials Register (eudraCT) and is a GCP (Good Clinical Practice) requirement.
For CTIMPS sponsored and/or co-sponsored by the University of Aberdeen and NHS Grampian, it is mandatory to use the Protocol template found on the SOP and Training page unless prior agreement from the sponsor has been approved.
When writing your protocol consider the following:
1) What is your specific question?
It needs to be relevant to other researchers in the field, practicing clinicians or patients.
2) What will be the best primary outcome to answer you question?
This needs to be observable, recordable and suitable for analysis.
3) Where and how will you recruit into the study?
Do you have access to enough participants to complete this study? Explain your recruitment plan.
4) Do you have enough sites?
To achieve the numbers required to make your study viable, will you need to make it multi centred.
5) What will be your Inclusion and Exclusion criteria?
This needs to be clear and free of any ambiguity.
6) How will Informed Consent be taken?
You will need to describe who, where, when and how informed consent is obtained and where the Informed Consent Forms will be kept
7) What is your plan for data collection? How will you ensure it is safe and how what will be your disaster recovery plan be?
Data collection (Case Report Forms) should be created in line with the protocol. You cannot collect more data than you need and you don't want to miss important measurements.
8) What will you do if a participant withdraws?
Be clear about whether you will be still keeping all data up to the point of withdrawal or how you are dealing with this for example. If you will replace withdrawn participants.
9) What safety measures do you have to put into place?
A robust system of safety reporting must be in place dependant on the risks of your study.
10) Compensation for participants?
Will you need to reimburse participants for travel time and/or inconvenience? Do you have funds for this?
11) Contracts/Agreements
Even if you are not paying for services and/or equipment you will still need a contract/agreement to create a clear record of the nature of the commitment (See next section).
Protocol Title
- The title is usually seen as the least important part of a study, but it can provide an extremely short synopsis of the trial in just one statement. It also supports internet searches if the key concepts of a study are mentioned in the title. Choose a title not by looking just at the acronym it could provide
- Important information (e.g. randomized, double-blind, parallel group) should be included in the title. Indexers on websites such as PubMed may not classify a report correctly if the authors do not explicitly report information in the title
Synopsis:
- A synopsis is a very helpful part of a protocol
- It should contain the key points of a study in tabular form
- It can act as quick reference when checking something and is a one-stop read for any newcomer
Statistical Analysis Plan (SAP)
- Take advice from a statistician!
- The SAP should not be an afterthought, but an essential part of a clinical study. It is recommended by the authorities and required by some journals now to ensure that data analysis has been performed in the right way.
- Remember that Protocols (and all other documents used in the study) should be version controlled and dated.